
Inzidenz und Prävalenz der AAV
Arten der AAV
Die am weitesten verbreiteten Subtypen der AAV sind Granulomatose mit Polyangiitis (GPA, zuvor bekannt als Morbus Wegener) und mikroskopische Polyangiitis (MPA). Ein weiterer Subtyp ist die eosinophile Granulomatose mit Polyangiitis (EGPA, zuvor bekannt als Churg-Strauss Syndrom).2,3 Diese Webseite befasst sich im Wesentlichen mit GPA und MPA.
AAV ist eine seltene Erkrankung2
- Globale Prävalenz: 30-218 pro Million4
- Inzidenz in Europa: 13-20 pro Million pro Jahr2,4
Auftreten der AAV
- AAV tritt sowohl bei jüngeren als auch bei älteren Menschen auf, jedoch nur selten bei Kindern und Jugendlichen. Mit zunehmendem Alter steigt die Inzidenz2,5
- AAV tritt bei Männern geringfügig häufiger auf (jährliche Inzidenzrate etwa 60 %) als bei Frauen2,5

Mögliche Organschäden
AAV kann eine Schädigung und ein Versagen systemischer Organe hervorrufen, wobei überwiegend die Nieren und die Lunge betroffen
sind1-3
AAV kann eine Schädigung lebenswichtiger Organe wie Lunge, Nieren, Nervensystem, Gastrointestinaltrakt, Haut, Augen und Herz hervorrufen.3,4 GPA und MPA zeigen zahlreiche klinische Gemeinsamkeiten, die EGPA unterscheidet sich dagegen deutlich.2
Bis zu 23 % der PR3- und MPO-ANCA-positiven Patienten mit RRT-Bedarf zum Zeitpunkt der Diagnose sterben innerhalb von 6 Monaten, und 29 % gewinnen die Nierenfunktion nicht zurück.5* Bei 13 % der PR3- und MPO-ANCA-positiven Patienten mit Beteiligung der Nieren zum Zeitpunkt der Diagnose entsteht innerhalb von 3 Jahren nach der Diagnose eine ESRD. Im Vergleich zu einem kurzen Prodromalstadium tritt bei Patienten mit langem Prodromalstadium (> 22 Wochen zwischen den ersten AAV-Symptomen und Diagnose) mit größerer Wahrscheinlichkeit nach 6 Monaten eine Proteinurie auf, und bei diesen Patienten besteht ein doppelt so hohes Risiko einer ESRD nach 3 Jahren. Dies unterstreicht den Stellenwert einer frühzeitigen Diagnose mit Blick auf eine Verbesserung der Nieren-Resultate.6†

GPA- und MPA-Patienten leiden zunehmend unter Organschäden, verursacht durch eine Kombination aus der Aktivität der Vaskulitis und glukokortikoidbedingten UE1–3
Langfristige und wiederholte Einnahme hochdosierter Glukokortikoide ist geknüpft an ein erhöhtes Risiko, an Diabetes mellitus zu erkranken bzw. ein bestehendes Krankheitsbild zu verschlechtern sowie Bluthochdruck, Osteoporose, avaskulärer Knochennekrose, Malignität, Katarakte und andere schwerwiegende Nebenwirkungen zu entwickeln oder zu verschlimmern.1,2*† In einer auf 7 Jahre angelegten Studie mit neu diagnostizierten GPA- und MPA-Patienten stieg die Schädigungshäufigkeit einschließlich potenziell therapiebedingter Schädigungen im Verlauf der Zeit (p<0,01). Nach der Diagnose kommt es bei den Patienten zunächst vermehrt zu Schäden, wobei 81,5 % der Patienten im VDI bei Monat 6 ≥1 Schäden entwickelt haben, verglichen zu 24,4 % der Patienten an der Baseline, wobei Nieren-(Proteinurie and GFR <50 mL/Minute) und Herz-Kreislaufschädigungen (Hypertonie) am häufigsten auftraten. Schwere Schädigungen mit einem VDI-Score von ≥5 nehmen im Lauf der Zeit zu, beim Langzeit-Follow-Up hatten 33,7 % aller Patienten schwere Schädigungen. Bluthochdruck war die am häufigsten berichtete Entwicklung zum Zeitpunkt des Langzeit-Follow-Up, ein Zeichen für die zunehmende Herz-Kreislaufschädigung im Lauf der Zeit. An der Baseline hatten 4,8 % der Patienten Bluthochdruck, innerhalb von 6 Monaten stieg dieser Anteil auf 17,0 %, und beim Langzeit-Follow-Up litten 41,5 % der Patienten an Hypertonie (p<0.01).2†
Schwere, langfristige Vaskulitisschäden wurden separat mit einer erhöhten kumulativen Glukokortikoid-Einnahme assoziiert (p = 0,016).3†
Diese Morbidität geht mit einem langfristig signifikant erhöhten Mortalitätsrisiko einher, mit einem Risikoquotienten von 2,41 (95 % KI: 1,74 - 3,34) bei GPA-Patienten im Vergleich zu Kontrollgruppen gleichen Alters und Geschlechts.2–4†‡
Durchschnittlich 7 Jahre nach der Diagnose für Patienten mit GPA oder MPA…2†

Mortalität und Morbidität
AAV führt zu einem erhöhten Mortalitätsrisiko, insbesondere im ersten Jahr nach der Diagnose1,2*
Im ersten Jahr nach der GPA- oder MPA-Diagnose (n = 524) liegt die Mortalitätsrate bei 10,7 % (n = 561)1*
Für diese Patienten gilt:
Die Hälfte (50 %) der Mortalität im ersten Jahr ist auf therapiebedingte Infektionen zurückzuführen1
In klinischen Studien lag die kumulative Überlebensrate nach 2 bzw. 5 Jahren bei Patienten mit neu diagnostizierter GPA oder und MPA bei 85 % bzw. 78 %.3†
Populationsbasierte Daten deuten darauf hin, dass die Langzeit-Mortalitätsrate bei AAV-Patienten in den letzten 20 Jahren erheblich gestiegen ist, jedoch noch nicht auf demselben Niveau wie bei vergleichbaren Kontrollpatienten liegt.2‡
In einer Studie mit Nachbeobachtung der Patienten über einen medianen Zeitraum von 5,2 Jahren war die Mortalitätsrate bei Patienten mit inzidenteller GPA oder MPA und konventioneller Behandlung 2,6-mal so hoch wie in der allgemeinen Population gleichen Alters und Geschlechts (p<0,0001).3†
Überweisung, Diagnose und Nachbeobachtung
AAV-Patienten durchlaufen häufig zahlreiche Etappen von der Überweisung bis zur Diagnose1*
Laut EULAR-Empfehlungen, benötigen Patient*innen Zugang zu Informationsmaterialien über die Auswirkungen von AAV und ihre Prognose, die wichtigsten Warnsymptome und die Behandlung (einschließlich behandlungsbedingte Komplikationen). AAV erfordert multidisziplinäres Behandlungsmanagement von Seiten der Versorgungszentren oder direkten Zugriff auf spezifisches Wissen über Vaskulitis.2

Viele Patient*innen haben bei der Vorstellung Nierenerkrankungen, aber unspezifische Symptome überwiegen, die zur Überweisung führen1
Nierenerkrankung:64%
Fatigue:58%
Fieber:54%
Gewichtsverlust:53%
Gelenkschmerzen:47%
16% der Patient*innen leiden länger als 3 Monate unter den Symptomen, die zur Überweisung führen, ehe eine AAV diagnostiziert wird1
Komorbiditäten sind zum Zeitpunkt der Diagnose häufig (65% der Patient*innen)1
Bluthochdruck:45%
Typ 2 diabetes:16%
COPD/Asthma:15%
Herzkranzerkrankungen:10%
Arthritis:9%
Osteoporose:7%
BMI >35:6%
Herzversagen:6%
Die relative Seltenheit und die unspezifischen Symptome der AAV kann die Diagnose bei einem Drittel der Patienten um mehr als 6 Monate verzögern.3
Die Diagnose der AAV und die Differenzierung in die Subtypen GPA, MPA oder EGPA ist abhängig von der Konstellation der klinischen Symptome des Patienten, von den Ergebnissen der bildgebenden Untersuchungen sowie von den Laboruntersuchungen.3,4
Angesichts der Assoziation von Anti-PR3 mit der GPA und Anti-MPO mit der MPA ist ein ANCA-Test für die Diagnose unerlässlich.3-7
- Bis zu 20 % der GPA- und MPA-Patienten sowie mehr als 60 % der EGPA-Patienten sind ANCA-negativ4
- Auch bei anderen Erkrankungen (z. B. Autoimmunhepatitis, Colitis ulcerosa, Hepatitis-C- oder HIV-Infektionen oder infektiöse Endokarditis) kann es zu einem positiven ANCA-Testergebnis kommen, ohne dass eine assoziierte Vaskulitis vorliegt4
Zur Bestätigung der Diagnose wird oft eine Organbiopsie (in der Regel eine Nierenbiopsie) vorgenommen.3,4
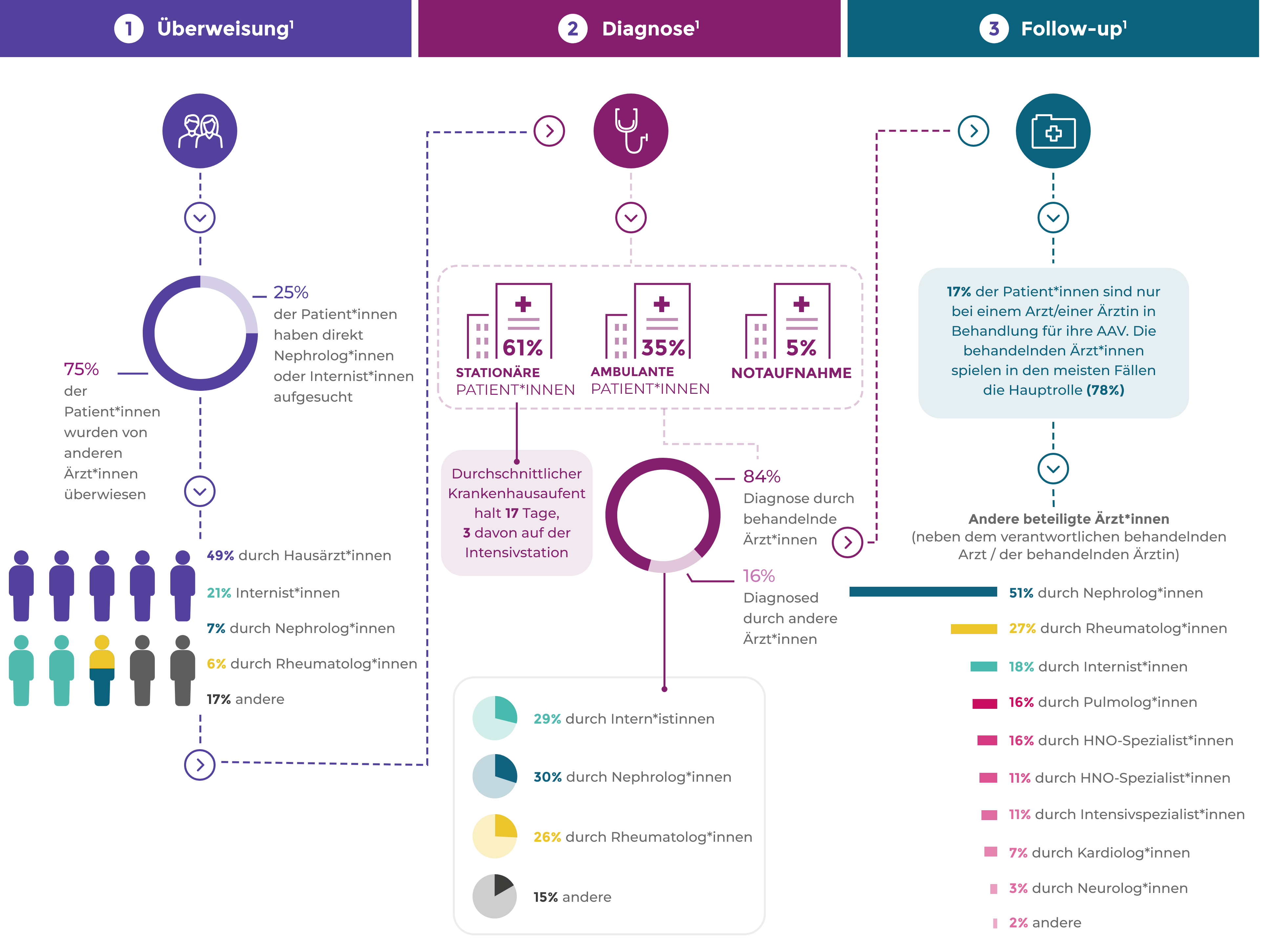
Überweisung1

Diagnose1

Follow-up1


Einführung in die AAV
AAV ist eine seltene, schwer verlaufende Vaskulitis der kleinen Blutgefäße mit Beteiligung mehrerer Organe und hohem Mortalitätsrisiko1
Weiterlesen
Krankheitsmechanismus
Die Interaktion zwischen dem aktivierten alternativen Komplementweg, den Neutrophilen und C5a steht im Zentrum der
vaskulitisbedingten
Schädigung durch die AAV2
Behandlung der AAV
Ein praktischer Leitfaden für die Behandlung und das Krankheitsmanagement von AAV-Patient*innen
Mehr erfahren










